Working with ugropy
Failing
![]()
ugropy may fail to obtain the subgroups of a molecule for a certain model for two reasons: either there is a bug in the code, or the molecule cannot be represented by the subgroups of the failing model.
ugropy utilizes SMARTS for the representation of functional groups to inquire whether the molecule contains those structures. Let’s examine the functional group list for the classic liquid-vapor UNIFAC model.
[1]:
try:
import google.colab
%pip install -q ugropy
except ImportError:
pass
[2]:
from ugropy import unifac
unifac.subgroups
[2]:
| smarts | molecular_weight | |
|---|---|---|
| group | ||
| CH3 | [CX4H3] | 15.03500 |
| CH2 | [CX4H2] | 14.02700 |
| CH | [CX4H] | 13.01900 |
| C | [CX4H0] | 12.01100 |
| CH2=CH | [CH2]=[CH] | 27.04600 |
| ... | ... | ... |
| NCO | [NX2H0]=[CX2H0]=[OX1H0] | 42.01700 |
| (CH2)2SU | [CH2]S(=O)(=O)[CH2] | 92.11620 |
| CH2CHSU | [CH2]S(=O)(=O)[CH] | 91.10840 |
| IMIDAZOL | [c]1:[c]:[n]:[c]:[n]:1 | 68.07820 |
| BTI | C(F)(F)(F)S(=O)(=O)[N-]S(=O)(=O)C(F)(F)F | 279.91784 |
113 rows × 2 columns
For example, let’s check the SMARTS representation of the alcohol group ACOH:
[3]:
unifac.subgroups.loc["ACOH", "smarts"]
[3]:
'[cH0][OH]'
The SMARTS representation it’s telling us that the OH group it’s, of course, a hydroxyl group bounded by a single bound to an aromatic carbon atom.
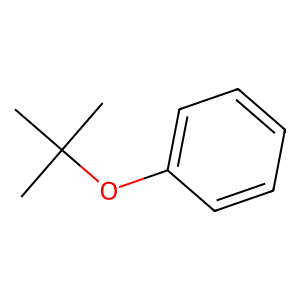
An example of a molecule that cannot be represented by UNIFAC groups:
[4]:
from rdkit.Chem import Draw
mol = unifac.get_groups("C1(=CC=CC=C1)OC(C)(C)C", "smiles")
Draw.MolToImage(mol.molecule)
[4]:
[5]:
print(mol.subgroups)
{}
The library “fails” to obtain any functional groups to accurately represent the molecule. This failure is represented by an empty dictionary. In this case, the “fail” is correct, but it could fail due to errors in the groups SMARTS representations or the algorithm, resulting in an empty dictionary as well. Currently, the supported models are tested against 444 different molecules.
If you encounter a failing representation, you can examine the structure of the molecule and the list of functional groups of the failing model. If you determine that the molecule can indeed be modeled, you may have discovered a bug. Feel free to report the issue on the repository along with the failing molecule’s SMILES/name, the failing model and the ugropy version.
More than one solution
Models like UNIFAC or PSRK can have multiple solutions to represent a molecule, and ugropy tries its best to find them all. In such cases, you will receive a list of dictionaries, each containing one of the solutions found. Let’s take a look.
[6]:
from ugropy import unifac
from rdkit.Chem import Draw
mol = unifac.get_groups("CCCC1=CC=C(CC(=O)OC)C=C1", "smiles", search_multiple_solutions=True)
Draw.MolToImage(mol[0].molecule, highlightAtoms=[7])
[6]:
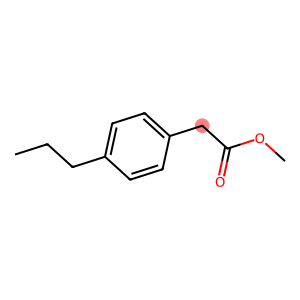
This molecule can be modeled in two ways depending on how we treat the CH2 attached to the ring and the ester carbon (highlighted in red). We can either form an ACCH2 group and model the ester group with COO, or we can use an AC group and model the ester group with CH2COO.
[7]:
print("Solution 1:")
print(mol[0].subgroups)
print("solution 2:")
print(mol[1].subgroups)
Solution 1:
{'CH3': 2, 'CH2': 1, 'ACH': 4, 'ACCH2': 2, 'COO': 1}
solution 2:
{'CH3': 2, 'CH2': 1, 'ACH': 4, 'AC': 1, 'ACCH2': 1, 'CH2COO': 1}
[8]:
mol[0].draw(width=800)
[8]:

[9]:
mol[1].draw(width=800)
[9]:

This could be useful in cases where some groups have more interaction parameters than others in the mixture that you want to model with UNIFAC. Alternatively, you can try both approaches and compare if there are any differences.
More solutions (non-optimal)
ugropy allows you to search multiple solutions for the models, but all these solutions are optimal (uses the same number of groups to represent the molecule). It’s possible to search for non-optimal solutions along with the optimal ones. This is useful when the user wants to find all possible combinations of fragments that cover the molecule. This feature is intended for research purposes and is not recommended to actually model the molecules.
We can do this with the search_nonoptimal parameter:
[10]:
from ugropy import unifac
solutions = unifac.get_groups(
"9,10-dihydroanthracene",
search_multiple_solutions=True, # If this is False, search_nonoptimal is ignored
search_nonoptimal=True,
)
solutions
[10]:
[<ugropy.core.frag_classes.gibbs_model.gibbs_result.GibbsFragmentationResult at 0x7f3e4d1a5cd0>,
<ugropy.core.frag_classes.gibbs_model.gibbs_result.GibbsFragmentationResult at 0x7f3e4d1a5ba0>,
<ugropy.core.frag_classes.gibbs_model.gibbs_result.GibbsFragmentationResult at 0x7f3e92cc3410>]
We can check all the obtained solutions:
[11]:
solutions[0].draw(width=600)
[11]:

[12]:
solutions[1].draw(width=600)
[12]:

[13]:
solutions[2].draw(width=600)
[13]:

If you are an experienced user of UNIFAC-like models, you may notice that the only “correct” solution is the first one. All ugropy models have the capacity to also search for non-optimal solutions.
Models information
You can check information about the models’ fragments. For example, the UNIFAC, PSRK, and Dortmund models have the information about the R and Q values, the subgroup number, and main group number. you can access to this information with model.subgroups_info:
[14]:
from ugropy import unifac, psrk, dortmund
unifac.subgroups_info
[14]:
| subgroup_number | main_group | R | Q | |
|---|---|---|---|---|
| group | ||||
| CH3 | 1 | 1 | 0.9011 | 0.848 |
| CH2 | 2 | 1 | 0.6744 | 0.540 |
| CH | 3 | 1 | 0.4469 | 0.228 |
| C | 4 | 1 | 0.2195 | 0.000 |
| CH2=CH | 5 | 2 | 1.3454 | 1.176 |
| ... | ... | ... | ... | ... |
| NCO | 109 | 51 | 1.0567 | 0.732 |
| (CH2)2SU | 118 | 55 | 2.6869 | 2.120 |
| CH2CHSU | 119 | 55 | 2.4595 | 1.808 |
| IMIDAZOL | 178 | 84 | 2.0260 | 0.868 |
| BTI | 179 | 85 | 5.7740 | 4.932 |
113 rows × 4 columns
Moreover, you can consult the fragments properties contributions of models like Joback:
[15]:
from ugropy import joback, abdulelah_gani
joback.properties_contributions
[15]:
| Tc | Pc | Vc | Tb | Tf | Hform | Gform | a | b | c | d | Hfusion | Hvap | na | nb | num_of_atoms | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| group | ||||||||||||||||
| -CH3 | 0.0141 | -0.0012 | 65.0 | 23.58 | -5.10 | -76.45 | -43.96 | 19.500 | -0.00808 | 0.000153 | -9.670000e-08 | 0.908 | 2.373 | 548.29 | -1.719 | 4 |
| -CH2- | 0.0189 | 0.0000 | 56.0 | 22.88 | 11.27 | -20.64 | 8.42 | -0.909 | 0.09500 | -0.000054 | 1.190000e-08 | 2.590 | 2.226 | 94.16 | -0.199 | 3 |
| >CH- | 0.0164 | 0.0020 | 41.0 | 21.74 | 12.64 | 29.89 | 58.36 | -23.000 | 0.20400 | -0.000265 | 1.200000e-07 | 0.749 | 1.691 | -322.15 | 1.187 | 2 |
| >C< | 0.0067 | 0.0043 | 27.0 | 18.25 | 46.43 | 82.23 | 116.02 | -66.200 | 0.42700 | -0.000641 | 3.010000e-07 | -1.460 | 0.636 | -573.56 | 2.307 | 1 |
| =CH2 | 0.0113 | -0.0028 | 56.0 | 18.18 | -4.32 | -9.63 | 3.77 | 23.600 | -0.03810 | 0.000172 | -1.030000e-07 | -0.473 | 1.724 | 495.01 | -1.539 | 3 |
| =CH- | 0.0129 | -0.0006 | 46.0 | 24.96 | 8.73 | 37.97 | 48.53 | -8.000 | 0.10500 | -0.000096 | 3.560000e-08 | 2.691 | 2.205 | 82.28 | -0.242 | 2 |
| =C< | 0.0117 | 0.0011 | 38.0 | 24.14 | 11.14 | 83.99 | 92.36 | -28.100 | 0.20800 | -0.000306 | 1.460000e-07 | 3.063 | 2.138 | NaN | NaN | 1 |
| =C= | 0.0026 | 0.0028 | 36.0 | 26.15 | 17.78 | 142.14 | 136.70 | 27.400 | -0.05570 | 0.000101 | -5.020000e-08 | 4.720 | 2.661 | NaN | NaN | 1 |
| CH | 0.0027 | -0.0008 | 46.0 | 9.20 | -11.18 | 79.30 | 77.71 | 24.500 | -0.02710 | 0.000111 | -6.780000e-08 | 2.322 | 1.155 | NaN | NaN | 2 |
| C | 0.0020 | 0.0016 | 37.0 | 27.38 | 64.32 | 115.51 | 109.82 | 7.870 | 0.02010 | -0.000008 | 1.390000e-09 | 4.151 | 3.302 | NaN | NaN | 1 |
| ring-CH2- | 0.0100 | 0.0025 | 48.0 | 27.15 | 7.75 | -26.80 | -3.68 | -6.030 | 0.08540 | -0.000008 | -1.800000e-08 | 0.490 | 2.398 | 307.53 | -0.798 | 3 |
| ring>CH- | 0.0122 | 0.0004 | 38.0 | 21.78 | 19.88 | 8.67 | 40.99 | -20.500 | 0.16200 | -0.000160 | 6.240000e-08 | 3.243 | 1.942 | -394.29 | 1.251 | 2 |
| ring>C< | 0.0042 | 0.0061 | 27.0 | 21.32 | 60.15 | 79.72 | 87.88 | -90.900 | 0.55700 | -0.000900 | 4.690000e-07 | -1.373 | 0.644 | NaN | NaN | 1 |
| ring=CH- | 0.0082 | 0.0011 | 41.0 | 26.73 | 8.13 | 2.09 | 11.30 | -2.140 | 0.05740 | -0.000002 | -1.590000e-08 | 1.101 | 2.544 | 259.65 | -0.702 | 2 |
| ring=C< | 0.0143 | 0.0008 | 32.0 | 31.01 | 37.02 | 46.43 | 54.05 | -8.250 | 0.10100 | -0.000142 | 6.780000e-08 | 2.394 | 3.059 | -245.74 | 0.912 | 1 |
| -F | 0.0111 | -0.0057 | 27.0 | -0.03 | -15.78 | -251.92 | -247.19 | 26.500 | -0.09130 | 0.000191 | -1.030000e-07 | 1.398 | -0.670 | NaN | NaN | 1 |
| -Cl | 0.0105 | -0.0049 | 58.0 | 38.13 | 13.55 | -71.55 | -64.31 | 33.300 | -0.09630 | 0.000187 | -9.960000e-08 | 2.515 | 4.532 | 625.45 | -1.814 | 1 |
| -Br | 0.0133 | 0.0057 | 71.0 | 66.86 | 43.43 | -29.48 | -38.06 | 28.600 | -0.06490 | 0.000136 | -7.450000e-08 | 3.603 | 6.582 | 738.91 | -2.038 | 1 |
| -I | 0.0068 | -0.0034 | 97.0 | 93.84 | 41.69 | 21.06 | 5.74 | 32.100 | -0.06410 | 0.000126 | -6.870000e-08 | 2.724 | 9.520 | 809.55 | -2.224 | 1 |
| -OH (alcohol) | 0.0741 | 0.0112 | 28.0 | 92.88 | 44.45 | -208.04 | -189.20 | 25.700 | -0.06910 | 0.000177 | -9.880000e-08 | 2.406 | 16.826 | 2173.72 | -5.057 | 2 |
| -OH (phenol) | 0.0240 | 0.0184 | -25.0 | 76.34 | 82.83 | -221.65 | -197.37 | -2.810 | 0.11100 | -0.000116 | 4.940000e-08 | 4.490 | 12.499 | 3018.17 | -7.314 | 2 |
| -O- (non-ring) | 0.0168 | 0.0015 | 18.0 | 22.42 | 22.23 | -132.22 | -105.00 | 25.500 | -0.06320 | 0.000111 | -5.480000e-08 | 1.188 | 2.410 | 122.09 | -0.386 | 1 |
| -O- (ring) | 0.0098 | 0.0048 | 13.0 | 31.22 | 23.05 | -138.16 | -98.22 | 12.200 | -0.01260 | 0.000060 | -3.860000e-08 | 5.879 | 4.682 | 440.24 | -0.953 | 1 |
| >C=O (non-ring) | 0.0380 | 0.0031 | 62.0 | 76.75 | 61.20 | -133.22 | -120.50 | 6.450 | 0.06700 | -0.000036 | 2.860000e-09 | 4.189 | 8.972 | 340.35 | -0.350 | 2 |
| >C=O (ring) | 0.0284 | 0.0028 | 55.0 | 94.97 | 75.97 | -164.50 | -126.27 | 30.400 | -0.08290 | 0.000236 | -1.310000e-07 | NaN | 6.645 | NaN | NaN | 2 |
| O=CH- (aldehyde) | 0.0379 | 0.0030 | 82.0 | 72.24 | 36.90 | -162.03 | -143.48 | 30.900 | -0.03360 | 0.000160 | -9.880000e-08 | 3.197 | 9.093 | 740.92 | -1.713 | 3 |
| -COOH (acid) | 0.0791 | 0.0077 | 89.0 | 169.09 | 155.50 | -426.72 | -387.87 | 24.100 | 0.04270 | 0.000080 | -6.870000e-08 | 11.051 | 19.537 | 1317.23 | -2.578 | 4 |
| -COO- (ester) | 0.0481 | 0.0005 | 82.0 | 81.10 | 53.60 | -337.92 | -301.95 | 24.500 | 0.04020 | 0.000040 | -4.520000e-08 | 6.959 | 9.633 | 483.88 | -0.966 | 3 |
| =O (other than above) | 0.0143 | 0.0101 | 36.0 | -10.50 | 2.08 | -247.61 | -250.83 | 6.820 | 0.01960 | 0.000013 | -1.780000e-08 | 3.624 | 5.909 | 675.24 | -1.340 | 1 |
| -NH2 | 0.0243 | 0.0109 | 38.0 | 73.23 | 66.89 | -22.02 | 14.07 | 26.900 | -0.04120 | 0.000164 | -9.760000e-08 | 3.515 | 10.788 | NaN | NaN | 3 |
| >NH (non-ring) | 0.0295 | 0.0077 | 35.0 | 50.17 | 52.66 | 53.47 | 89.39 | -1.210 | 0.07620 | -0.000049 | 1.050000e-08 | 5.009 | 6.436 | NaN | NaN | 2 |
| >NH (ring) | 0.0130 | 0.0114 | 29.0 | 52.82 | 101.51 | 31.65 | 75.61 | 11.800 | -0.02300 | 0.000107 | -6.280000e-08 | 7.490 | 6.930 | NaN | NaN | 2 |
| >N- (non-ring) | 0.0169 | 0.0074 | 9.0 | 11.74 | 48.84 | 123.34 | 163.16 | -31.100 | 0.22700 | -0.000320 | 1.460000e-07 | 4.703 | 1.896 | NaN | NaN | 1 |
| -N= (non-ring) | 0.0255 | -0.0099 | NaN | 74.60 | NaN | 23.61 | NaN | NaN | NaN | NaN | NaN | NaN | 3.335 | NaN | NaN | 1 |
| -N= (ring) | 0.0085 | 0.0076 | 34.0 | 57.55 | 68.40 | 55.52 | 79.93 | 8.830 | -0.00384 | 0.000044 | -2.600000e-08 | 3.649 | 6.528 | NaN | NaN | 1 |
| =NH | NaN | NaN | NaN | 83.08 | 68.91 | 93.70 | 119.66 | 5.690 | -0.00412 | 0.000128 | -8.880000e-08 | NaN | 12.169 | NaN | NaN | 2 |
| -CN | 0.0496 | -0.0101 | 91.0 | 125.66 | 59.89 | 88.43 | 89.22 | 36.500 | -0.07330 | 0.000184 | -1.030000e-07 | 2.414 | 12.851 | NaN | NaN | 2 |
| -NO2 | 0.0437 | 0.0064 | 91.0 | 152.54 | 127.24 | -66.57 | -16.83 | 25.900 | -0.00374 | 0.000129 | -8.880000e-08 | 9.679 | 16.738 | NaN | NaN | 3 |
| -SH | 0.0031 | 0.0084 | 63.0 | 63.56 | 20.09 | -17.33 | -22.99 | 35.300 | -0.07580 | 0.000185 | -1.030000e-07 | 2.360 | 6.884 | NaN | NaN | 2 |
| -S- (non-ring) | 0.0119 | 0.0049 | 54.0 | 68.78 | 34.40 | 41.87 | 33.12 | 19.600 | -0.00561 | 0.000040 | -2.760000e-08 | 4.130 | 6.817 | NaN | NaN | 1 |
| -S- (ring) | 0.0019 | 0.0051 | 38.0 | 52.10 | 79.93 | 39.10 | 27.76 | 16.700 | 0.00481 | 0.000028 | -2.110000e-08 | 1.557 | 5.984 | NaN | NaN | 1 |
From all fragmentation models you have all the RDKit mol objects of each fragment already instantiated on a dictionary. This could be helpful to for calculations, multiple solution filtering, or visualization:
[16]:
unifac.detection_mols["CH3"]
[16]:

[17]:
unifac.detection_mols["CH2O"].GetNumAtoms()
[17]:
2
Changing the default ILP solver
By default, ugropy uses the default CBC solver from the PulP library. But PulP supports a lot of different solvers. An example could be de XPRESS_PY solver.
First, you must install XPRESS with:
pip install xpress
Then you can change the default solver with a simple argument:
[18]:
from ugropy import unifac
unifac.get_groups("toluene", solver_arguments={"solver": "XPRESS_PY"}).draw(width=600)
/opt/hostedtoolcache/Python/3.13.7/x64/lib/python3.13/site-packages/pulp/apis/xpress_api.py:751: LicenseWarning: Using the Community license in this session. If you have a full Xpress license, pass the full path to your license file to xpress.init(). If you want to use the FICO Community license and no longer want to see this message, use the following code before using the xpress module:
xpress.init('/opt/hostedtoolcache/Python/3.13.7/x64/lib/python3.13/site-packages/xpress/license/community-xpauth.xpr')
model = xpress.problem()
[18]:

Changing the ILP solver has impact on the execution time of the library.